

Reading Time: 4 minutesHi! My name is Katie and I have recently joined the MND Association’s Research team as a Research Insight Co-ordinator. I’ll be working to stay

Reading Time: 5 minutesHello! Libby here, one of our Research Impact Specialists at the MND Association. Before joining the Association, I completed my PhD in MND research at

Reading Time: 5 minutesAround one in ten people diagnosed with MND have a family history of the disease. Some people also have a family history of a related

Reading Time: 5 minutesDiscovering new treatments for everyone with MND matters and clinical trials play a vital role in making that happen. The success of Tofersen, an effective
36th Symposium: San Diego Causes and disease mechanisms Events Genetic Research Markers of Disease Progression MND Research Symposium
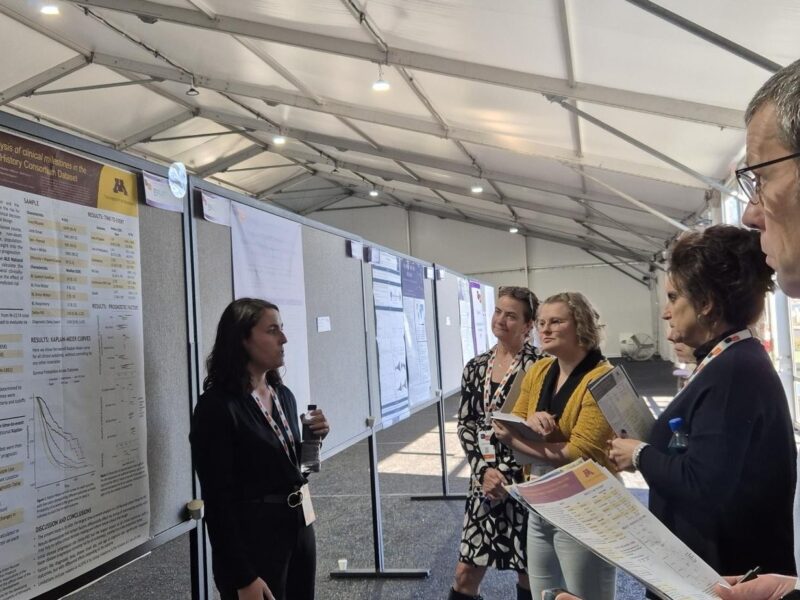
Catching up with the 2024 Poster prize candidates
Reading Time: 6 minutesEvery year, hundreds of research projects from around the world are showcased at the International Symposium, reflecting the sheer amount of research going on in

36th Symposium: San Diego Causes and disease mechanisms Healthcare MND Research Models of MND Symposium
Celebrating Early Career MND Researchers through the International Symposium on ALS/MND Poster Prize
Reading Time: 5 minutesEach year at the International Symposium, we recognise early career researchers whose work is making exceptional contributions to understanding and treating MND. This prestigious award acknowledges

Reading Time: 5 minutesPrimary Lateral Sclerosis (PLS) is a type of MND that progress slowly over many years. It is estimated that around 3 in 100 people diagnosed

Reading Time: 6 minutesThis year, we are welcoming our 11th cohort of Non-Clinical Research Fellows to our incredible team of more than 320 scientists involved in research grants

36th Symposium: San Diego Causes and disease mechanisms Events Markers of Disease Progression MND Research Models of MND News Symposium
Bringing the community together for the 36th International Symposium on ALS/MND
Reading Time: 4 minutesThe International Symposium on ALS/MND has closed its doors once again after three insightful, inspiring and hopeful days! After being postponed in 2020 due to the COVID-19 pandemic, we finally made

Reading Time: 2 minutesThe International Symposium on ALS/MND has closed its doors after three insightful, inspiring and hopeful days! This year’s event in San Diego really has showcased the breadth of research happening around the