My name is Oscar Wilkins and I am a PhD student working with Professors Pietro Fratta (recipient of a MND Association Research Fellowship) and Jernej Ule. Recently, in collaboration with Sarah Hill and Michael Ward, a team led by PhD student Anna-Leigh Brown, postdoctoral researcher Matt Keuss and myself uncovered a promising new target for MND therapies. You can read the full paper in Nature. In parallel, a separate Stanford-based group led by Aaron Gitler independently came to the same conclusions, meaning the study has already been reproduced! This study has also been published in the latest issue of Nature.
Relocation, Relocation
Proteins are small molecular machines that perform many essential functions within cells. Arguably the most important protein in MND research is TDP-43, as, in almost all MND cases, TDP-43 is incorrectly ejected from the cell’s nucleus (where the DNA is stored), and instead found in the surrounding cytoplasm. This has two big implications for neuronal health: on the one hand it means that TDP-43 cannot perform its important functions within the cell’s nucleus (a “nuclear loss of function”), and on the other hand it might cause problems in the cytoplasm (a “cytoplasmic gain of function”).
In our study, we investigated the former: how the loss of TDP-43 function in the nucleus impacts neuronal health. To examine this, we grew neuronal cells, derived from human stem cells, in dishes, then removed the TDP-43 protein from these cells using a new technology based on CRISPR-Cas9. We then studied how these neurons without TDP-43 differed from healthy neurons.
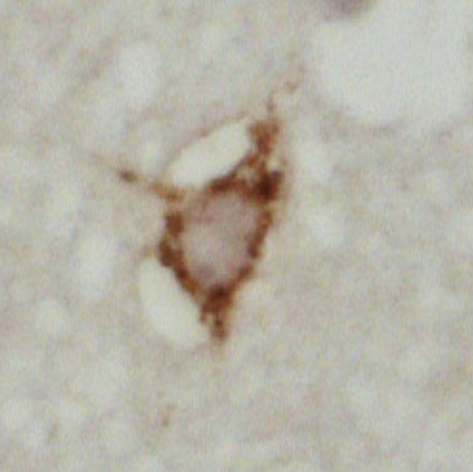
Figure 1 – showing mislocalisation of TDP-43 in ALS motor cortex cells (top), compared to healthy motor cortex cells (bottom)
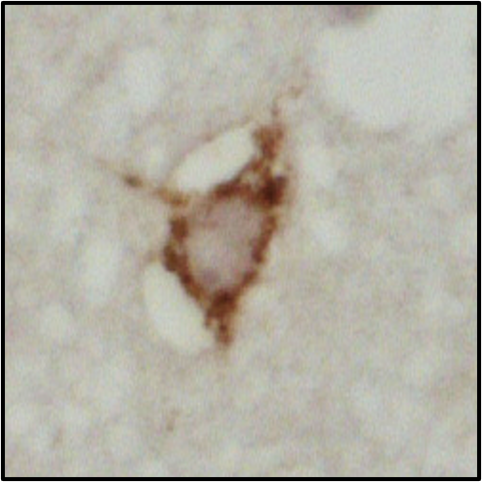
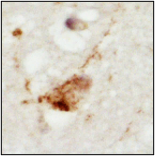
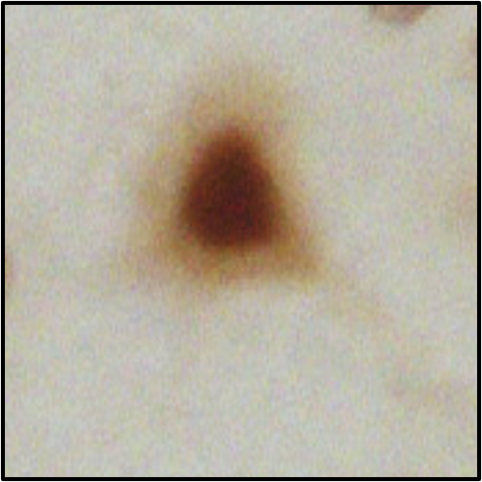
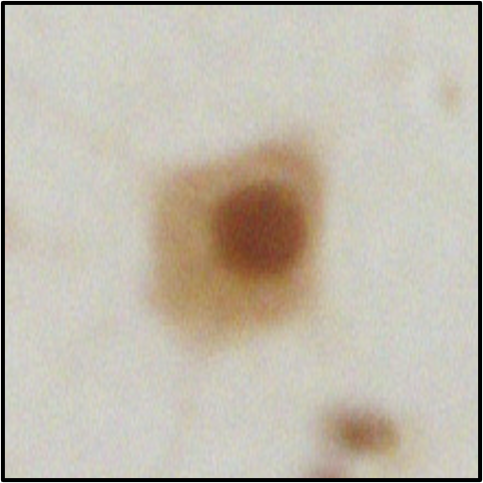
Figure 2 – showing mislocalisation of TDP-43 in ALS lower motor neurons (top), compared to healthy lower motor neurons (bottom)
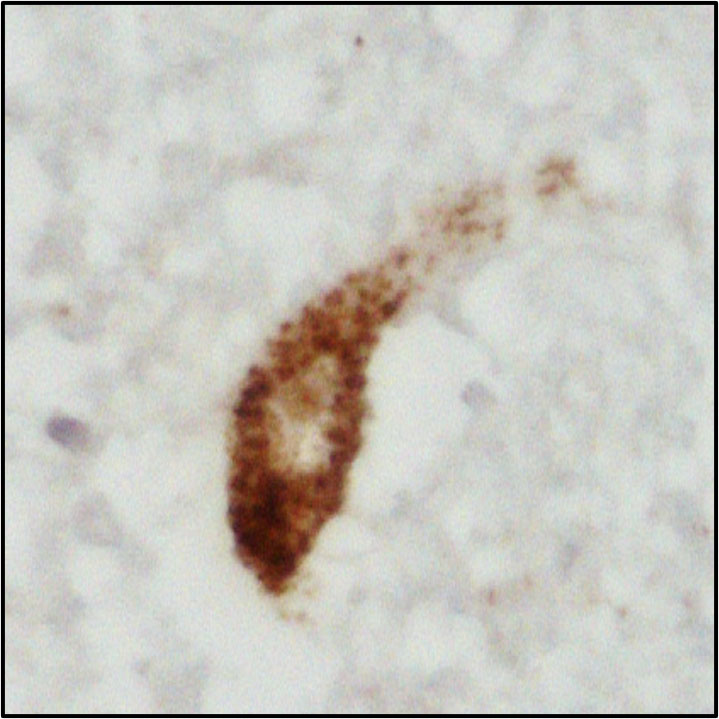
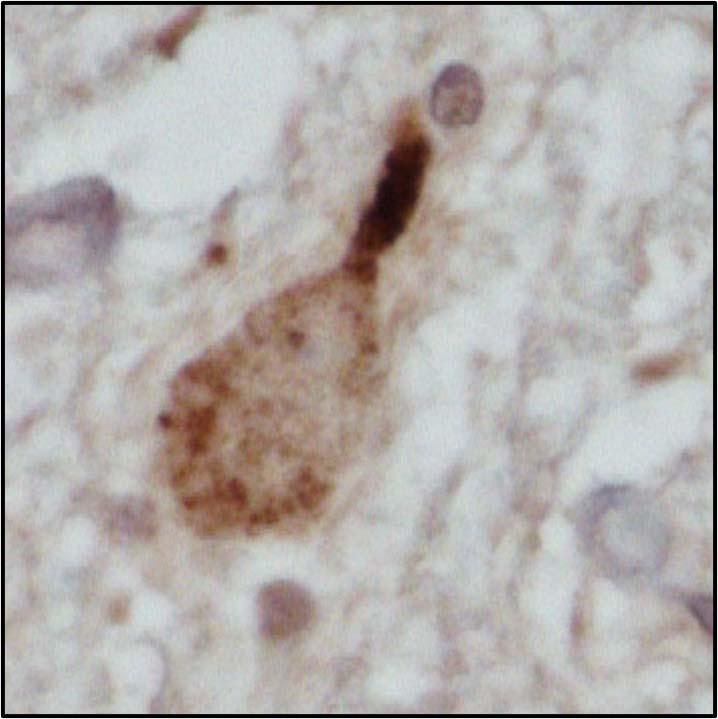
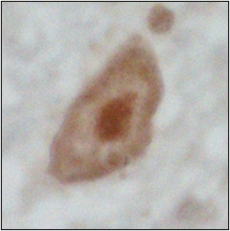
Figures 1 and 2 explained
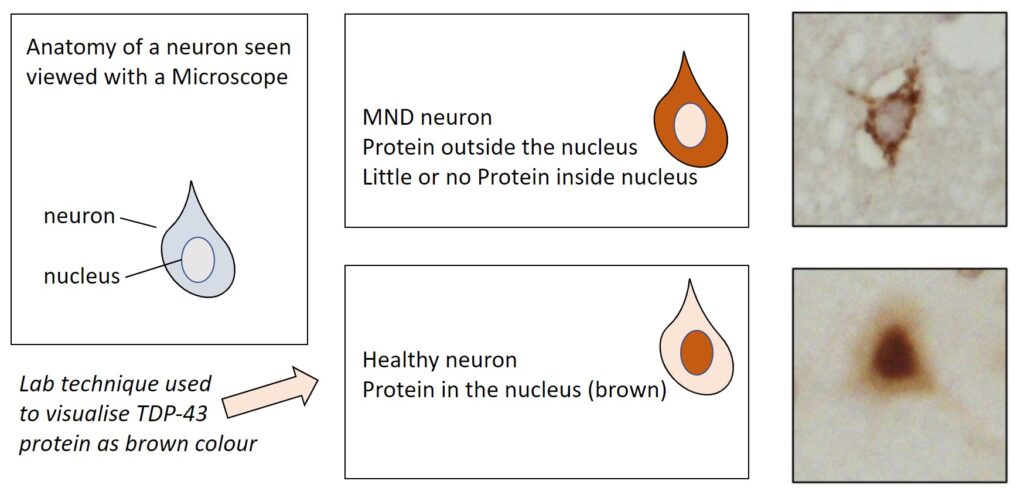
Photos courtesy of Alex Bampton and Tammaryn Lashley, co-authors of the paper
A corrupt messenger
DNA contains instructions for how to make the ~20,000 different proteins found in the human body. To make a protein, the instructions for this single protein are first copied from the DNA to a small piece of RNA, called messenger RNA (mRNA). This mRNA is then sent to a “protein factory” called a ribosome, which reads the instructions and builds the protein. It’s like the DNA is a cookbook, the mRNA is a single photocopied recipe, and the ribosome is the chef. Of course, it’s essential that the instructions from the DNA are copied correctly…
Strikingly, when we examined the mRNA molecules in the neurons we had generated, we found that the mRNAs for a protein called UNC13A were corrupted, meaning the ribosomes in the neurons were unable to correctly produce the UNC13A protein. Furthermore, when we looked at patient samples, we again found that the mRNAs for UNC13A were incorrect.
This finding jumped out at us for two reasons. First, UNC13A is one of the most important proteins for neuronal function, as it enables neurons to communicate with each other via neurotransmitter release. The corruption of the UNC13A mRNAs in patients is thus likely to impair neuronal function and contribute to neurodegeneration.
Second, UNC13A genetic variants are among the strongest genetic risk factors for MND and frontotemporal dementia, but the reason for this has remained a mystery. We hypothesised that these genetic variants might increase the risk of the UNC13A mRNAs being corrupted. Excitingly, when we tested this theory in neuron-like cells grown in dishes, and in patient samples, we indeed found that the genetic risk variants substantially increased the level of UNC13A mRNA corruption.
From the laboratory to the clinic
These results are a significant breakthrough for several reasons. On a purely scientific level, they explain why UNC13A genetic variants increase risk of MND and dementia, a question which has puzzled researchers for over a decade. They are also the first to demonstrate a genetic link specifically between loss of nuclear TDP-43 function and MND, improving our understanding of this central disease mechanism.
Excitingly, these results also point to a promising new therapy for MND and frontotemporal dementia. By using small DNA-like molecules, it is likely we can stop the UNC13A mRNAs from being corrupted. Similar approaches have already been applied to spinal muscular atrophy (SMA) and Duchenne muscular dystrophy (DMD). We are hopeful that such an approach could slow the progression of MND and frontotemporal dementia. It will be some time before new treatments based on our findings can enter the clinic, but we have already started work on this, so fingers crossed!